An epidemic may be seen as a whole, an entity to be studied, which we can refer to as a process. This process results from underlying mechanisms, which we can refer to as sub-processes. Therefore, the building blocks of plant disease epidemics, as processes, consist of sub-processes. For instance, for an aerially dispersed disease, one may consider the following sub-processes: propagule production, propagule liberation, propagule transport, propagule deposition, infection, latency period, and infectious period. The process itself thus consists of linked sub-processes, monocycle components, which have been collectively called the ‘infection chain’ by J. Kranz (1974). It is the concatenation of infection chains that leads to an epidemic. This chapter deals with the modeling of an epidemic, as a process, on the basis of knowledge of processes at the next lower level of integration, that is to say, the monocycle components.
One could also model one of the sub-processes, such as propagule formation (for example for an asexually reproducing fungus: conidiophore initiation, conidiophore elongation, conidiophore branching, spore initiation, spore maturation). One could also consider the system where epidemics are sub-processes of a higher-scale process called polyetic epidemics. In this case, it is the concatenation of successive individual epidemics, as lower levels of integration, which results in polyetic epidemics occurring over many successive seasons, as upper levels of integration. The former case might be of interest to study, e.g., how genes involved in spore production in a pathogen influence the dynamics of sporulation, which in turn can have applications in understanding one of the biological bases of host plant resistance. The latter case is of interest in understanding how, and to what extent, successive epidemics are related (especially via the primary inoculum), i.e., what is the basis of the carry-over of epidemics across time, and this can have applications in disease management over seasons.
Biology, in general, is concerned with hierarchies of processes. It is up to the scientist to choose which level of this hierarchy should be the focus of an investigation. This chapter focuses on epidemics. Epidemics, as biological phenomena, can be decomposed in sub-processes, which in turn can be decomposed in sub-sub-processes. Systems analysis, in turn, using (among other tools) simulation modeling, enables one to investigate and understand the behavior of one level of a system's hierarchy, making use of knowledge acquired on the next-lower level of integration within a biological hierarchy.
This chapter concentrates on one of today's principal structures in plant disease epidemiology. This structure remains an important, quite current (e.g., Cunniffe et al., 2012; Van den Bosch et al., 2008; Segarra et al., 2001), field of investigation in its own, although having been published by Zadoks in 1971 (Zadoks, 1971). The model elaborates on the foundations developed by Van der Plank (1963), with the concepts of:
- infection,
- latency period, and
- infectious period,
which are captured by the differential-delay equation:
dxt /
dt = Rc (xt-p -
xt-i-p) (1 -
xt) (equation 8.3, p. 100, Van der Plank, 1963),
where
xt is the amount of disease at time
t, Rc is the basic infection rate corrected for removals,
p is the latency period duration, and
i is the infectious period duration.
Components of a preliminary epidemiological model
The simulation model developed by Zadoks (1971) provides a numerical integration of Van der Plank's seminal equation of botanical epidemiology. As in the earlier example, we need to first define the system under consideration and its components.
The system under consideration is a 1-m2 crop area surrounded by similar crop areas. This crop consists of sites, which may be healthy (HSites), or infected. Sites that have been infected can be partitioned in three, non-overlapping, categories: sites that have been infected but are not yet infectious, and therefore are latent (LatS), sites that are infectious and are therefore generating propagules (InfS), and sites that are no longer infectious and thus are removed from the infectious process (RemS). The notion of site, therefore, refers to those plant tissues that can sustain a given infection and give rise to new ones. Sites, therefore, will not be the same depending on the pathosystem: for example, in the case of systemic diseases, a site will refer to an entire plant unit, whereas for leaf- (or, e.g., fruit-) spotting diseases, a site will refer to a (potential or existing) lesion.
In the following, let us concentrate on the case of a disease that is aerially dispersed and causes lesions on leaves. This is an important consideration, because it determines the nature of the state variables that are of primary concern in the considered system. In this case, we are thus dealing with a population of sites, whose transitions from healthy, to latent, to infectious, and to removed, are dynamically tracked.
In addition to the choice of system's limits, let us further assume that the time step is one day. Many epidemiological models use such a time step, in large part because, for most weather data sets available, the climatic day starts at about 7 a.m., and ends the following day at the same time. In the intervening time ― a full day ― many epidemiological events happen in our system: for instance, spores are produced, liberated, and deposited, and infections take place. Epidemiologists are well aware that these events do depend on environmental factors (the weather, but possibly the physiological status of the host plants, too) that vary with a much smaller time constant (e.g., a wind gust in the canopy, a short shower, or the progressive dry-off of moist leaves). However, these factors may only influence mechanisms that are themselves sub-processes of a sub-process (i.e., the infection process) at hand. In other words, these factors influence sub-sub-processes, whereas our endeavor is to numerically integrate sub-processes and quantify their consequences at the process level. Yet, one must devise a modeling approach that enables to consider sub-processes themselves, without ignoring the possible importance of processes at a lower level of integration. This is directly related to the time constant we assume the system has. We shall return to this important consideration at the end of this chapter.
Main equations of the model
Since the sites are non-overlapping categories, one may write:
ACI = LatS+InfS+RemS
where ACI is the total number of infected sites. However, only infectious sites (InfS) can produce propagules which can lead to additional InfS, while infection can only take place on sites that are still healthy at one point of time. Thus, it is convenient to calculate:
CORF = 1-(ACI/(ACI+HSites))
where CORF, a 'correction factor' for site availability, represents the proportion of healthy sites that are still available for infection, or the probability of a site to be healthy (CORF is strictly comprised between 0 and 1). Being a ratio of quantities with the same dimensions ([N]), the dimension of CORF is: [N.N-1] ≡ [1].
In the course of any day, propagules are being produced, released, transported, and deposited, and infection may take place on healthy sites. The rate of infection can be expressed as:
INFECTION = DMFR*CORF*InfS
In this equation, INFECTION is a rate, and therefore has the dimension of a speed: [N.T-1]. InfS is the number of infectious sites at a given point of time ([N]), CORF is the correction factor, and DMFR is a daily multiplication factor (Daily Multiplication FactoR). DMFR is the number of daily new infections originating from existing, infectious lesions, or, the daily number of daughter-lesions per mother-lesion, with dimension [N.N-1.T-1]. In essence, DMFR is analogous to the intrinsic rate of bacterial population increase of the previous chapter. From an epidemiological standpoint, DMFR, in the mechanistic wording used in this model, exactly corresponds to Rc, the basic infection rate corrected by removals used by Van der Plank (1963). The dimension of INFECTION is therefore [N.N-1.T-1].[1].[N] ≡ [N.T-1]. It has the dimension of a rate, that is, of the speed of a process.
Note that the INFECTION equation also collapses a number of sub-sub-processes together, some of which were mentioned above. In other words, with respect to the daily rate of infection, all of which happens in any given day is summarized in the equation for INFECTION. This, of course, is a very important simplification, which we make in order to adhere to the above principle of systems analysis: explaining a process from its immediately related sub-processes. This simplification can, however, be further documented while remaining at the same levels of integration. This will be revisited later on.
Additional elements need to be documented in the model structure. Both the latency period and the infectious period are important phases of the disease monocycle. Sites that are in both the latent and the infectious stages correspond to two state variables. These state variables, however, are of a particular type, because sites remain in these states for specified (and epidemiologically important) durations. Such delays in a given state are called residence times. Thus, we want to assign a residence time in these two states,
p days in the latent, and
i days in the infectious stage. These have been called 'boxcar trains' (to reflect a series of boxes through which each individual progresses; Penning de Vries and Van Laar, 1982), or, using the terminology used in STELLA®, 'conveyors'. We need to decide what these residence times are. Let us, for a start, assume that
p = 6 days and
i = 10 days.
Let us also further assume for the sake of simplicity that values for
p and
i are fixed throughout the duration of an epidemic. Epidemiologists know that this is a strong simplification: for instance, as plants become older both
p and
i may vary, expressing increasing, or decreasing, resistance with development. Both parameters are also bound to change during the course of an epidemic with weather variables. Simulation modeling (and software such as STELLA® in particular) enables one to incorporate daily changes in values of
p and
i from driving variables, such as the crop development stage or varying temperature. For now, because we want to first develop a simple model structure, let us nevertheless assume that both parameters do not change. Let us further assume that, as the epidemic starts, both state variables contain no individuals, that is, that there are no infected sites in the latent or the infectious stages.
Initializing the model
A number of statements need to be made in order to run the model. First one needs to specify the population size of the host. Let us assume that the initial number of healthy sites (HSites) is 100,000. Let us further assume that the duration of an epidemic is 100 days.
Next, we need to define a value for the daily multiplication factor, DMFR. Let us, for a start, assume that DMFR = 0.3. A value of 0.3 for DMFR means that every day, a (mother) infectious site (InfS) can potentially give rise to 0.3 (daughter) infected site through the INFECTION rate. 'Potentially' implies here that there is enough 'space' for 0.3*InfS new infections to take place, that is, that propagules will reach healthy sites (HSites). In so doing, we only express the underlying hypotheses of Van der Plank's (1963) equation. We know that there are many constraints on the occurrence of infection besides the availability of healthy sites. We shall re-visit this key assumption at the very end of this chapter.
Another element concerns the initialization of the epidemic. There are several ways to do so. One approach could be to place infected sites in the latent stage as a starting point. Instead, because we want to be able to vary the date at which the epidemic starts, let us create an INOCPRIM parameter representing the amount of primary inoculum, which becomes active at a chosen point of time in the course of the growing season, and let us create a connected DAY parameter, which simply tracks time in the model (note that under STELLA®, 'TIME' represents the elapsing computing time). Let us further decide for now that INOCPRIM generates a single influx of new infections through INFECTION. We thus create a starting device, written as:
INFECTION = (DMFR*CORF*InfS) + INOCPRIM
DAY = TIME
INOCPRIM = IF (DAY=1) THEN 100 ELSE 0
which states that at a given day (here, day 1), there is a single, one-day, influx of active primary inoculum (INOCPRIM) resulting in 100 sites becoming infected (latent).
Drawing the model's flow chart
Our model flow chart is shown in Fig. 4.1, and depicts the different state variables and parameters, as well as their relationships. Note that the state variables for latent and infectious sites are shown as conveyors, and also the series of links determining the rate of infection (INFECTION), which reflect the series of assumptions made on state variables and parameters that determine its daily value.
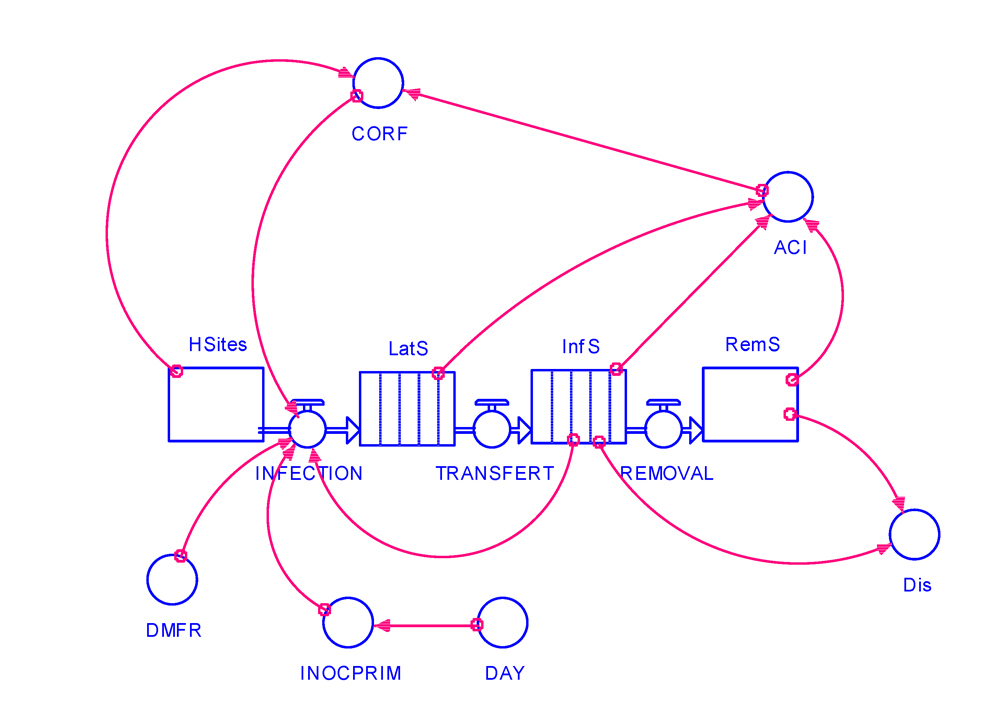
Figure 4.1. Flowchart of a preliminary epidemiological model for an aerially dispersed pathogen. HSites: healthy sites; LatS: latent sites; InfS: infectious sites; RemS: removed sites; Dis: (visibly) diseased sites; ACI: accumulated infected sites; CORF: correction factor for site infection; DMFR: daily multiplication factor; INOCPRIM: primary inoculum; DAY: running day. See Figure 2.1 and Table 2.1 for the meaning of symbols.
Model verification: a first run
A first stage in model evaluation consists of checking whether the model's program executes the intended instructions as originally designed (Penning de Vries and Van Laar, 1982). Such a task is easy in the case of this preliminary epidemiological model, and even easier in the case of the bacterial population model discussed in the previous chapter. Fig. 4.2 gives a graphical output of the dynamics of an epidemic. We can make the following remarks:
- the amount of visibly infected sites (Dis = InfS+RemS; curve 5) increases in a sigmoid pattern, as the stock of healthy sites is being depleted, and the effect of CORF on INFECTION comes into effect, slowing down the speed of the epidemic;
- the amount of latent lesions (curve 3) progressively increases and then decreases, representing, in many ways, the slope of the disease (Dis) progress curve;
- the amount of infectious sites (curve 2) follows the same pattern as the amount of latent lesions, with a delay of about 6 days, that is, as expected, about
p;
- removed sites (curve 4) accumulate regularly, as infected sites exit the latent and infectious stages.
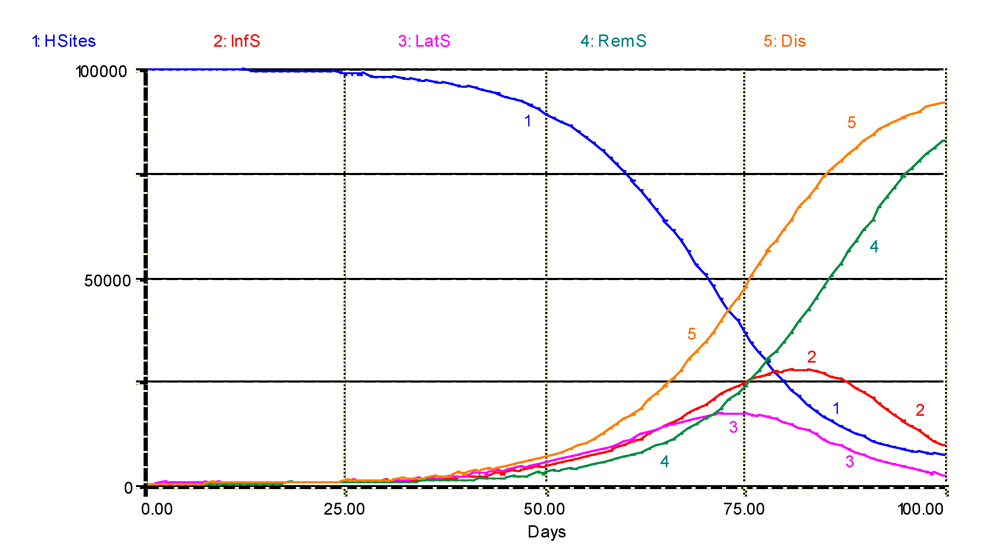
Figure 4.2. First output of a preliminary simulation: healthy, latent, infectious, removed, and visibly diseased sites. The values of parameters used are DMFR = 0.3 lesions.lesion-1.day-1, p = 6 days, i = 10 days, and date of onset is 1. 1: healthy sites; 2: infectious sites; 3: latent sites; 4: sites removed from the epidemiological process; 5: accumulated visibly diseased sites (infectious and removed). Horizontal axis: time (days); vertical axis: numbers of sites.
The overall behavior of the model is, therefore, as expected and shows the patterns of disease progress described in so many reports. It does, therefore, conform to the expected. This model provides a visual and quantitative solution to the equation developed by Van der Plank some 50 years ago (simulation outputs could also be displayed in a tabular manner, which is not shown here).
A key element that simulation modeling brings about is the possibility to see what is not visible: Fig. 4.2 displays the dynamics of latent lesions, which, of course, would be impossible to monitor in the field. This same remark applies to removed and infectious sites which, in nearly all cases would be impossible to tell apart even for the most experienced field pathologist. Simulation modeling, therefore, allows visualization of the behavior not only of the process, but of the sub-processes considered.
Exploring the model's behavior
The effects of variations of a series of parameters in the model are shown in Fig. 4.3. This sensitivity analysis can be summarized as follows: (1) increasing values of DMFR from 0.01 to 0.5 lesion·lesion·day-1 strongly increases the speed of epidemics (Fig. 4.3a); (2) increasing values of
p from 1 to 12 days strongly suppresses epidemics (Fig. 4.3b); (3) increasing values of
i from 1 to 20 days strongly increases the final level of disease (Fig. 4.3c); and (4) delaying the epidemic from 1 to 30 days strongly reduces the final level of disease as well (Fig. 4.3d).
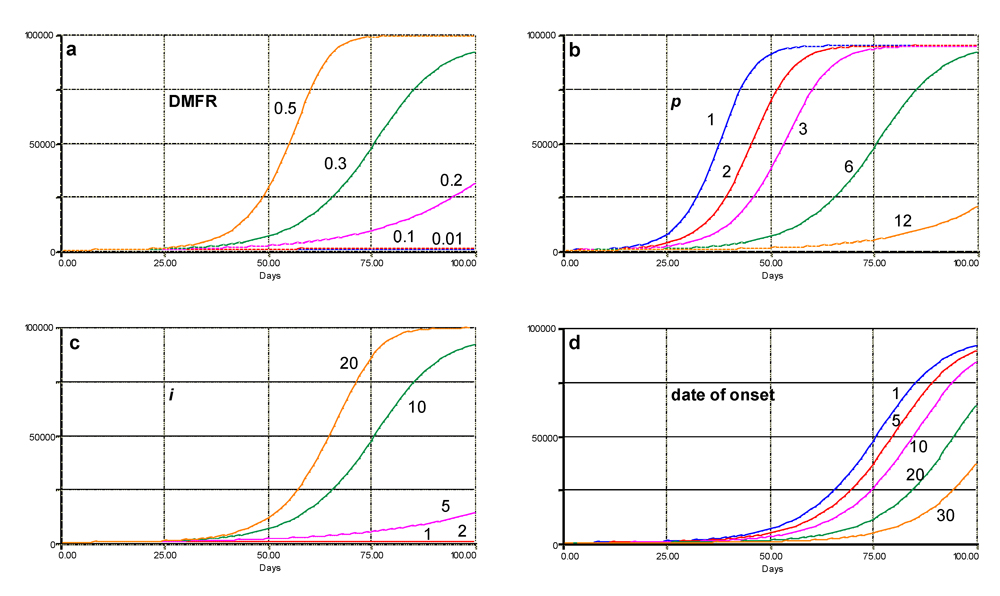
Figure 4.3. Sensitivity analyses of variations in DMFR, latency period, infectious period, and date of epidemic onset. The simulated number of diseased sites (infectious and removed) is displayed in all figures. a: effects of varying DMFR values; b: effects of varying values of latency period; c: effects of varying values of infectious period; d: effects of varying dates of onset values. The default parameters values are DMFR = 0.3 lesions.lesion-1.day-1, p = 6 days, i = 10 days, and date of onset is 1. Values of parameters which are varied are indicated on the simulated curves. Horizontal axis: time (days); vertical axis: numbers of sites.
These are well-known effects of key epidemiological parameters (Van der Plank, 1963; Zadoks, 1971; Zadoks and Schein, 1979) on plant disease epidemics involving a large number of overlapping and concatenated infection chains. This further indicates that the model structure we developed conforms to what has become to be known as classical epidemiological theory.
Another outcome of these runs is, simply, that simulation modeling allows one to "see" Kranz's (1974) infection chain in action in a polycyclic process. Often, changes in value of a parameter has little immediate effect, but, as disease cycles overlap in the course of an unfolding epidemic (Teng, 1983), the compounding effect becomes stronger, sometimes with dramatic consequences.
Revisiting hypotheses
The development of this model structure, despite its conforming to known epidemiological principles, is based on a number of hypotheses. Exploring these hypotheses are grounds for very current and active research (e.g., Segarra et al., 2001; Cunniffe et al., 2012). We address some of these hypotheses briefly, with a focus on assessing the validity of the model structure.
A first hypothesis concerns the area of the system considered and its boundaries. The system under consideration here consists of a 1-m2 crop area surrounded by similar systems. A 1-m2 crop area may, for instance, be relevant for a cereal or a legume crop. One would obviously have to increase this size when considering most perennials or semi-perennial crops. The assumption also implies that these boundaries allow fluxes of propagules to enter and exit the system in a steady-state. What is implied by 'similar crop areas' surrounding our system is that the amount of disease is the same in surrounding areas. It also implies that the crop structure does not vary greatly, so that the microclimatic conditions would, in our system, be representative of the conditions that prevail in neighboring, equivalent systems. In this preliminary epidemiological example, the system under consideration is kept as simple as possible; the limited size of the system, the variability of the host population size over time, the consequences the host population size may have on microclimate or disease spread, for instance, are disregarded for the sake of simplicity. The hypothesis of a limited system surrounded by similar systems with which it is in a dynamic equilibrium (e.g., flows of propagules, heat, water vapor) is often referred as the 'mean field' hypothesis. Although the model may have usefulness in its ability to understand and compare quantitatively sub-processes (components of the epidemiological monocycle) in determining the outcome of processes (epidemics), the 'mean field' is a very strong hypothesis which must lead to cautious interpretation of results.
Another hypothesis is that we refer here to aerially-dispersed (fungal or bacterial) diseases. This has two implications; one is that the model structure deals with processes that are typical of aerial dispersal. The other implication is that the population of sites which is considered consists of fractions of leaf tissues that potentially may become lesions. Regarding this second implication, one should note that the same model structure can effectively be used to address sites of other dimensions in a host population hierarchy, from fractions of leaves, to leaves, to shoots, or entire plants (Savary et al., 2012).
A further assumption in the model is that we chose to use a one-day time step. This leads us to collapse the liberation-transport-deposition-infection process into a single rate, INFECTION, governed by DMFR and CORF. One must question whether DMFR could possibly be kept constant throughout a cropping season. DMFR can actually be made dependent on time, whether because weather varies (and thus affects dispersal and infection sub-sub-processes) or host tissues become less and less susceptible over time as they age. In the case of weather variation, driving functions (e.g., air temperature) could be entered in the model as tabulated values using STELLA® and Excel®. Simple assumptions can also be made. In the case of ageing, and increasingly resistant, tissues, let us for instance assume that DMFR decreases exponentially as:
DMFR = 0.3*EXP(-k*DAY)
where k is a positive extinction coefficient. The outputs of three values for k (0: no resistance, 0.005: moderate resistance, and 0.01: strong resistance) are plotted in Fig. 4.4, showing again how DMFR strongly influences the behavior of the model.
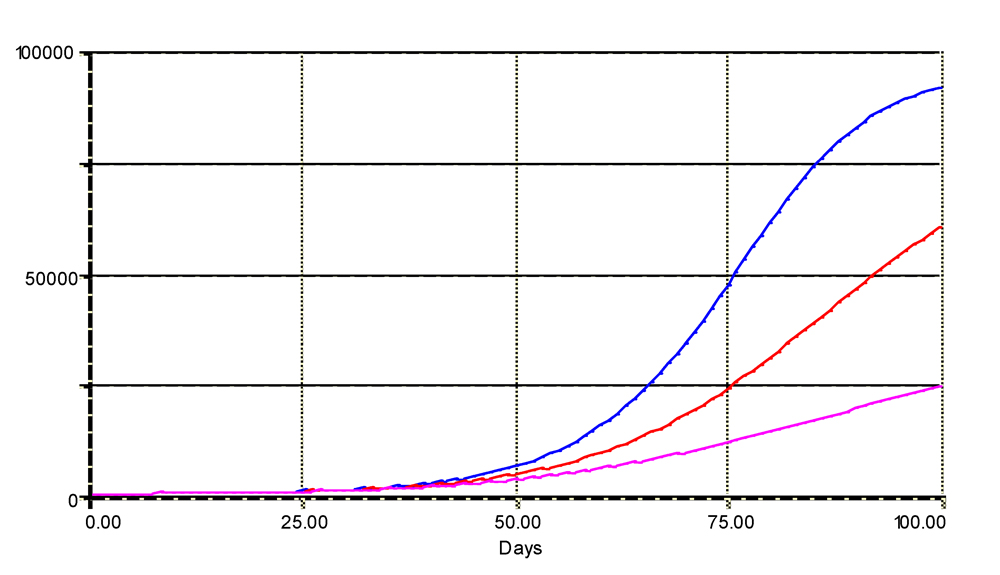
Figure 4.4. Simulated disease progress with exponentially decreasing values of DMFR.
The simulated number of diseased sites (infectious and removed) is displayed.
Upper, medium, and lower curves are simulated with an extinction coefficient over time of 0, 0.01, and 0.005, respectively. See text for details. Horizontal axis: time (days); vertical axis: numbers of sites.
Similar approaches could be used if one wanted to address other sub-sub-processes, such as spore germination, germ tube elongation, penetration, and establishment of host-pathogen interaction, leading to actual infection.
Conversely, one can hardly imagine that, in the real world, both the latency,
p, and infectious,
i, periods would remain constant throughout a 100-day epidemic (e.g., Cunniffe et al., 2012). The current model architecture is flexible enough to incorporate such changes. For instance, the TRANSFERT rate between the two boxcars representing the latency and the infectious periods can be made also a function of temperature (in addition to the inherent characteristics allowing such state variables to vary properly; de Wit and Goudriaan, 1978). Let us assume that, for instance, during the 100 days, the environmental conditions (say, an increasing temperature) translate into having increasing values of
p. This can be simulated, and the simulation outputs are shown in Fig. 4.5. The increasing value of
p leads, as expected, to a slower disease progress, and to a lower final disease intensity.
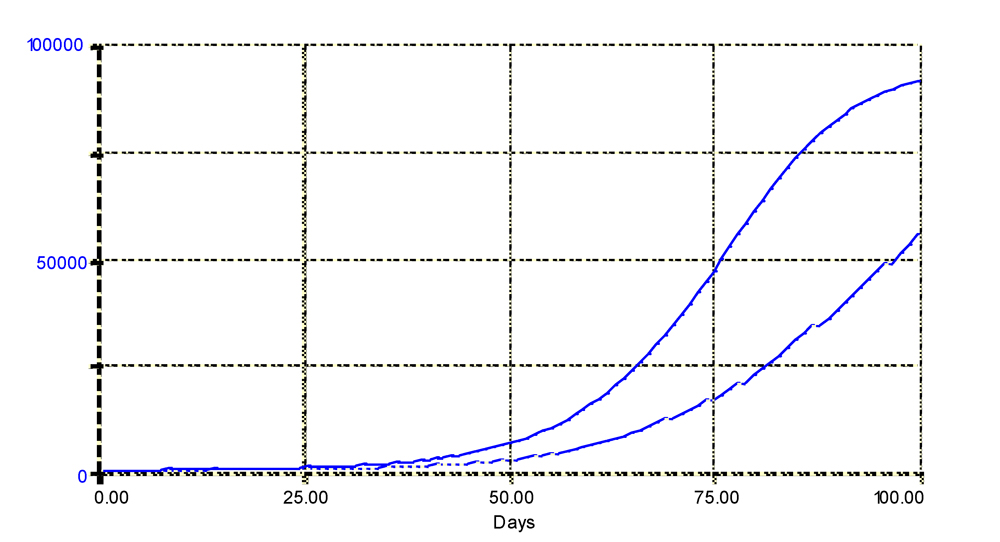
Figure 4.5. Simulated disease progress curves with constant (upper curve) or varying (lower curve) temperature influencing the latency period duration, p. The lower curve is associated with days when temperature may be high, leading to an increase in p. The simulated number of diseased sites (infectious and removed) is displayed. Horizontal axis: time (days); vertical axis: numbers of sites.
The preliminary model in a broader context (1): the basic infection rate corrected for removals
Let us look backward, and consider again Van der Plank's differential-delay equation:
dxt /
dt = Rc (xt-p -
xt-i-p) (1 -
xt),
and let us remember that the preliminary simulation model discussed here provides a numerical integration of this equation. Two key differences between analytical and numerical integration can be highlighted. While analytically solved equations produce exact solutions, numerical integration over a chosen time interval, Δt, only generates numerical estimates, which however can easily be derived, even when parameters vary over time. Simulation modeling provides an easy way to integrate Van der Plank's equation, which is quite complicated analytically (Madden et al., 2007).
The preliminary model in a broader context (2): the basic reproduction number
The total number of newly infected individuals resulting from a single infected individual occurring in a totally healthy population has been referred to as the basic reproduction number (or ratio),
R0 (Diekmann et al., 1990).
R0 has been extensively used in human and animal epidemiology (e.g., Molisson, 1995). In botanical epidemiology,
R0 is the progeny-parent ratio, or the number of daughter lesions per mother lesion, when the mother lesion is established in a population of healthy individuals (Van den Bosch et al., 1988a; Zadoks and Schein, 1979; Van der Plank, 1963). The concept of
R0 has recently been the subject of strong interest in botanical epidemiology, under the mathematical framework of linked differential equations (e.g., Segarra et al., 2001; Madden et al., 2007).
The preliminary model in a broader context (3): linking Rc and R0
Considering the infectious ― the reproductive ― life-time, that is, from
t = 0 to
t =
i, of the first infection being established in a population of sites that are all susceptible, one may write:
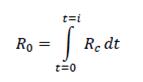
R0 (also called gross reproduction) has also been shown to be an important parameter to consider when analyzing disease focus expansion (Van den Bosch et al., 1988a; 1988b). Links between developments in medical and botanical epidemiology in terms of
R0 and
Rc were discussed later on in several studies (e.g., Jeger and Van den Bosch, 1993).
R0 is a very appealing concept, because of its clear definition, its biological meaning, its possible decomposition in biological processes, and because it is an important factor determining epidemics.
R0 is, however, very difficult to estimate (Van den Bosch et al., 2008b). Several approaches have been offered, including:
- deriving equations relating
r, the apparent rate of infection, or
rl, the logarithmic infection rate (sensu Van der Plank, 1963) to
R0 or
Rc (Van der Plank, 1963; Van den Bosch et al., 1988b; Sun and Zeng, 1994; Segarra et al., 2001);
- using matrix population models (Van den Bosch et al., 2008);
- experimental measurement (Van den Bosch et al., 1988); and
- approaches combining experiments and models (Allorent et al., 2005).
One way to see the preliminary epidemiological simulation model described here is that it allows the computation of the product of DMFR by
i, at successive, discrete time steps. This product in turn corresponds to
R0. R0 encompasses the entire infectious lifetime of a lesion, whereas
Rc considers each (infinitely small) time step over time during the infectious period. Thus, in the same way as
Rc,
R0 varies over time and depends on weather variables, e.g., temperature and leaf wetness (e.g., Papastamati and Van den Bosch, 2007; Zadoks and Schein, 1979).
Van der Plank (1963) stated that no plant disease epidemic can start unless
Rc⋅i > 1, an inequality known to epidemiologists as 'the threshold theorem'. This inequality simply states that if an infectious site does not give rise to a new infection, then no epidemic would take place. Much work has elaborated on the threshold theorem (Madden et al., 2007).
The mean field hypothesis
Whichever approach is chosen, whether analytical or numerical, the model considered here is based on the major assumption that all healthy sites are equally accessible for infection, or that, conversely, propagules all have equal probabilities to reach and (possibly) infect healthy sites. This is what may be called a mean field hypothesis. Canopies are, in the real world, heterogeneous; leaves or fruits, or plant organs ― sites, in general ― are not all equally exposed to incoming inoculum; tissues vary in their susceptibility; gradients of propagule dispersal vary widely across pathosystems, and these gradients often depend on several, not one, dispersal mechanisms; or again, the microclimate in a canopy (say, in an apple tree, but in a wheat or rice field, too) is bound to show spatial variability. All these elements, which may explain why an epidemic occurs, or why it does not, are left aside at this stage. What has been shown in this chapter truly is a preliminary model. Simulation modeling is one approach that enables to explore further the 'what if' questions that we have.
Simulations
The STELLA® model provided with this chapter (EPIDEM.STMX) will allow you to explore the model structure and equations, and run the model with varying values of DMFR,
p,
i, and onset date of epidemics, to see the effects of such changes on the simulated epidemics. A listing of the program can be found in Appendix 4.1.
Summary
This chapter shows and assembles the building blocks of a preliminary epidemiological simulation model.
- The main equations governing the model are explained.
- Ways to initialize the model are shown.
- The model behaves as expected, providing a verification of its structure and its equations.
- The model behaves as expected from pathosystems that correspond to its structure: variation in some key parameters (e.g., the durations of the latency and infectious periods, the intrinsic rate of disease increase - called here the daily multiplication factor, and the date of epidemic onset) translate in logical epidemiological patterns.
- Simulation modeling allows one to "see" the infection chain in action in a polycyclic process, with the compounding effects of parameter values influencing overlapping disease cycles of an epidemic.
- The many simplifying hypotheses of the model are discussed.
- The model structure developed in this chapter is discussed with respect to other current modeling approaches.
- A STELLA® model provided with this chapter (EPIDEM.STMX) can be used to see the effects of parameter changes on the simulated epidemics.
References
Allorent, D., Willocquet, L., Sartorato, A., and Savary, S. 2005. Quantifying and modelling the mobilisation of inoculum from diseased leaves and defoliated tissues in epidemics of angular leaf spot of bean. Eur. J. Plant Pathol. 113:377-394.
Brown, L. R. 2011. World on the Edge - How to Prevent Environmental and Economic Collapse. Earth Policy Institute. W.W. Norton & Company, New York, London.
Cunniffe, N. J., Stutt, R. O. J. H., Van den Bosch, F., and Gilligan, C. A. 2012. Time-dependent infectivity and flexible latent and infectious periods in compartmental models of plant disease. Phytopathology 102:365-380.
Diekmann, O., Heesterbeek, J. A. P., and Metz, J. A. J. 1990. On the definition and computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations. J. Math. Biol. 28:365-382.
Jeger, M. J., and Van den Bosch, F. 1993. Threshold criteria for model plant disease epidemics. I. Asymptotic results. Phytopathology 84:24-27.
Kranz, J., ed. 1974. Epidemics of plant diseases: Mathematical Analysis and Modeling. Springer-Verlag, Berlin.
Madden, L. V., Hughes, G., and Van den Bosch, F. 2007. The Study of Plant Disease Epidemics. American Phytopatholological Society, St. Paul, MN.
Mollison, D., ed. 1995. Epidemic Models: their Structure and Relation to Data. Cambridge University Press, Cambridge, UK.
Papastamati, K., and Van den Bosch, F. 2007. The sensitivity of the epidemic growth rate to weather variables, with an application to yellow rust on wheat. Phytopathology 97:202-210.
Penning de Vries, F. W. T., and Van Laar, H. H., eds. 1982. Simulation of Plant Growth and Crop Production. Pudoc, Wageningen.
Savary S., Nelson A., Willocquet L., Pangga I., and Aunario J., 2012. Modelling and mapping potential epidemics of rice diseases globally. Crop Protection 34:6-17.
Segarra, J., Jeger, M. J., and Van den Bosch, F. 2001. Epidemic dynamics and patterns of plant diseases. Phytopathology 91:1001-1010.
Sun, P., and Zeng, S. 1994. On the measurement of the corrected basic infection rate. Zeitschrift für Pflanzenkrankheiten und Pflanzenschurtz 101:297-302.
Teng, P. S. 1983. Estimating and interpreting disease intensity and loss in commercial fields. Phytopathology 73:1587-1590.
Van den Bosch F, Zadoks, J. C., and Metz, J. A. J. 1988a. Focus expansion in plant disease. I. The constant rate of focus expansion. Phytopathology 78:54-58.
Van den Bosch F, Frinking, H. D., Metz, J. A. J., and Zadoks, J. C. 1988b. Focus expansion in plant disease. III: two experimental examples. Phytopathology 78:919-925.
Van den Bosch, F., McRoberts, N., Van den Berg, F., and Madden, L. V. 2008. The basic reproduction number of plant pathogens: Matrix approaches to complex dynamics. Phytopathology 98:239-249.
Van der Plank, J. E. 1963. Plant Diseases. Epidemics and Control. Academic Press, New York.
de Wit, C. T., and Goudriaan, J. G. 1978. Simulation of Ecological Processes. Pudoc, Wageningen.
Zadoks, J. C. 1971. Systems analysis and the dynamics of epidemics. Phytopathology 61:600-610.
Zadoks, J. C., and Schein, R. D. 1979. Epidemiology and Plant Disease Management. Oxford University Press, New York.
Suggested reading
Allen, T. H. F., and Starr, T. B. 1982. Hierarchy – Perspectives for Ecological Complexity. The University of Chicago Press, Chicago and London.
Kranz, J. 1990. Epidemics, their mathematical analysis and modeling: an introduction. Pages 1-11 in: Epidemics of Plant Diseases. Second Edition. Kranz, J., Ed. Springer Verlag, Berlin.
Exercises and questions
Questions
1. A sub-process is
- 1 hierarchy level below the process level
- 2 hierarchy levels below the process level
- 1 hierarchy level above the process level
- 2 hierarchy levels above the process level
2. State variables of an epidemiological simulation model can include
- the rate of infection
- the duration of latency period
- the number of latent sites
- the correction factor
3. The rate of infection in an epidemiological simulation model can be written as
a. RG = RemS * DMFR
b. RG = (DMFR*CORF*InfS) + INOCPRIM
c. RG = (DMFR* INOCPRIM) + (CORF*InfS)
d. RG = (CORF*InfS) + INOCPRIM
4. The dimension of the rate of infection is
a. [N]
b. [N.T-1]
c. [N.T-2]
d. [N.N-1.T-1]
5. The following state variables may have an initial value set to zero
a. number of healthy sites
b. number of latent sites
c. number of infectious sites
d. number of removed sites
Answers to questions
1. a: 1 hierarchy level below the process level.
2. c: the number of latent sites.
3. b: RG = (DMFR*CORF*InfS) + INOCPRIM
4. b: [N.T-1]
5. b: number of latent sites, c: number of infectious sites, and d: number of removed sites.
Appendix 4.1. Program listing of EPIDEM
HSites(t) = HSites(t - dt) + (- INFECTION) * dt
INIT HSites = 100000
OUTFLOWS:
INFECTION = (DMFR*CORF*InfS)+INOCPRIM
InfS(t) = InfS(t - dt) + (TRANSFERT - REMOVAL) * dt
INIT InfS = 0,0,0,0,0,0,0,0,0,0
TRANSIT TIME = 10
INFLOW LIMIT = INF
CAPACITY = INF
INFLOWS:
TRANSFERT = CONVEYOR OUTFLOW
OUTFLOWS:
REMOVAL = CONVEYOR OUTFLOW
LatS(t) = LatS(t - dt) + (INFECTION - TRANSFERT) * dt
INIT LatS = 0,0,0,0,0,0
TRANSIT TIME = 6
INFLOW LIMIT = INF
CAPACITY = INF
INFLOWS:
INFECTION = (DMFR*CORF*InfS)+INOCPRIM
OUTFLOWS:
TRANSFERT = CONVEYOR OUTFLOW
RemS(t) = RemS(t - dt) + (REMOVAL) * dt
INIT RemS = 0
INFLOWS:
REMOVAL = CONVEYOR OUTFLOW
ACI = LatS+InfS+RemS
CORF = 1-(ACI/(ACI+HSites))
DAY = TIME
Dis = InfS+RemS
DMFR = 0.3
INOCPRIM = IF (DAY=1) THEN 100 ELSE 0