Gene flow is a unifying force that prevents populations from diverging. Gene flow breaks down the geographical or other boundaries that could otherwise isolate populations. As a result of isolation between populations, and the consequent limitations in exchange of genes, we expect that populations will diverge by genetic drift or as a result of selection for alleles that adapt each population to its local niche. But if gene flow occurs at a sufficiently high level, then otherwise isolated populations will not diverge genetically. Instead, they become united and evolve as a single evolutionary unit. Gene flow is especially important for plant pathogens in agroecosystems because it is the process that introduces new genes into agricultural fields distant from the site of the original mutation. This process is probably important in natural ecosystems as well. Gene flow moves virulence alleles into new populations. Gene flow thus introduces new alleles that can displace old alleles, if they are better adapted to the current host.
In populations which are made up of one or a few clonal lineages, a special case of gene flow can occur in which each clone (i.e. a genotype) has several mutations that differentiate it from the dominant, pre-existing clone. Given the fact that many genes move together as a block in asexual clones, it is better to think of "genotype flow." Genotype flow then refers to the movement of entire genotypes (usually clones or clonal lineages) between distinct populations. Genotype flow occurs only for organisms that have a significant asexual component to their life cycle. As an example, genotype flow occurs when a genotype (clone) of Fusarium oxysporum f. sp. melonis (cause of Fusarium wilt on melons) moves from North America to Israel on the muddy boots of an agricultural scientist. In this case, F. oxysporum does not have a sexual cycle, so the entire set of alleles in the clone is introduced into a new population. If this clone has a high degree of fitness, it can become established in the new location. Though recombination is possible for bacteria and viruses, it is reasonable to consider these pathogens as exhibiting mainly genotype flow, while fungi can exhibit a mixture of gene and genotype flow.
The population subdivision that results from genetic drift can be overcome by gene flow. The easiest model to consider how this process works is the Continent-Island model proposed by the population geneticist Sewall Wright. The following example will illustrate this model.
Assume that a is the virulent mutant allele that occurs at an avirulence locus (and A is the corresponding avirulence allele). Assume further that the frequency of the mutant a allele is q. Represent the frequency of the a allele as f(a) and the frequency of the A allele is f(A).
Let f(a) = q and let f(A) = p

Figure 6. The continent-island model assumes that gene flow occurs in only one direction, from a donor population (continent) to a recipient population (island).
Let:
m = the proportion of the island population that consists of migrants
1-m = the proportion of the island population that consists of natives
Q = the frequency of the virulence allele a in the "donor" (continent) population
qo = the frequency of the virulence allele a in the "recipient" (island) population
After one cycle of gene flow, we find that:
q1 = (1-m)qo + mQ
![]() q = -m(qo - Q)
q = -m(qo - Q)
where ![]() q = q1-q0
q = q1-q0
This formula can be used to calculate how fast allele frequencies will change through gene flow. As an example, let’s consider the hypothetical movement of a virulence allele for leaf rust from the UK to France.
f(a) = 0.50, the UK population has a high frequency of the virulence allele because most UK wheats have Lr13 R-gene.
qo = 0.00,
m = 0.05, the migration rate is high because a large number of spores were deposited by a migration event caused by a wind storm moving spores across the English Channel.
![]() q = -0.05(0.00-0.50) = 0.025
q = -0.05(0.00-0.50) = 0.025
q1 = 0.025 ~3% of the French population now contains avrLr13
At equilibrium (after many cycles of gene flow driven by many storms sweeping across the English Channel), allele frequencies of the donor and recipient populations become the same, qo = Q. So the frequency of avrLr13 will go to 0.50 in France even if French wheat breeders never use Lr13 in their resistant wheat cultivars. This is one possible explanation for the unexpected high frequency of virulence alleles in populations of some pathogens when the host population lacks the corresponding resistance gene (Bousset et al. 2002; Caffier et al. 1996; Hovmoller 2001).
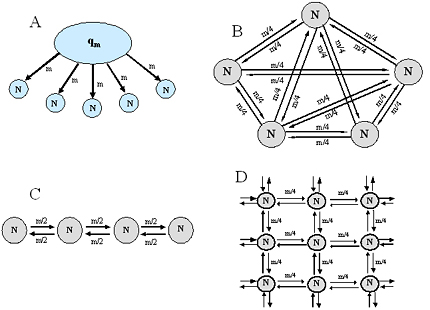
Many other models of gene flow have been described in addition to the island model. Figure 7 shows examples of one- and two-dimensional stepping-stone models and more complex multidimensional models of gene flow. Each of these models represents a permutation of the same scheme and can be adapted to the reality of the agricultural or natural ecosystem under study.
 |
|
Figure 7. Illustration of different models of gene flow. A) Continent-island model; B) Full island model; C) One-dimensional stepping stone model; and D) Two-dimensional stepping stone model. Click here to see an enlargement of this figure. |
The end result of gene flow is to make populations become genetically similar. This is illustrated in Figure 8, which shows how quickly geographically separated populations converge on the same allele frequency when 10% of each population is made up of immigrants from the other populations.
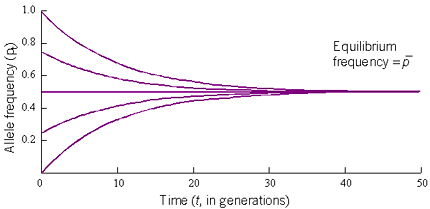 |
| Figure 8. Change of allele frequency against time in five subpopulations exchanging migrants at the rate m=0.1 per generation. Note the rapid convergence to a common equilibrium frequency. |
Several good examples of long-distance regional and global gene flow exist for fungal pathogens in agricultural ecosystems.
 |
|
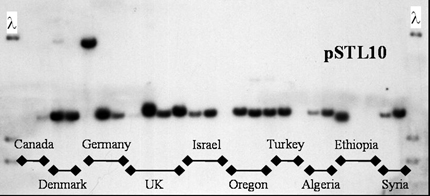
Figure 9. Shared RFLP alleles at the pSTL10 RFLP locus in Mycosphaerella graminicola populations from 3 continents.
|
Example 1: Evidence for global gene flow among populations of the wheat leaf blotch pathogen Mycosphaerella graminicola (anamorph Septoria tritici). RFLP (restriction fragment length polymorphism) alleles are shared between populations around the world (Figure 9) and allele frequencies are remarkably similar among populations on different continents (Table 2). But no isolates with shared DNA fingerprints were found in different populations (Zhan et al. 2003). This shows that the individual genotype that moved to a new population did not persist, but its genes were passed into the recipient population through its sexual offspring. The greatest gene diversity was found in the population from Israel, which is the center of origin of the wheat host (Zhan et al. 2003). The center of diversity for the pathogen suggests that this also is the center of origin of M. graminicola. This fits the standard model for gene flow. Zhan et al. (2003) hypothesized that ascospores disseminate genes over distances of 100s of km, while infected seeds disseminate genes between continents.
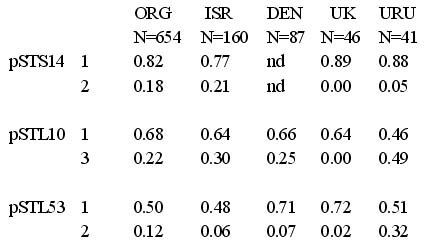 |
| Table 2. Similarity in RFLP allele frequencies at three RFLP loci among Mycosphaerella graminicola populations from Oregon, Israel, Denmark, United Kingdom, and Uruguay. N refers to the number of isolates assayed in each population |
Example 2: Evidence for regional genotype flow for the banana wilt pathogen Fusarium oxysporum f. sp. cubense, which causes Panama disease
DNA fingerprints detected the same genotypes in different countries (Koenig et al. 1997; Bentley et al. 1998). This fungus probably moves regionally and between plantations on infected banana cuttings that are used to start new plantations. The greatest genotypic diversity in the pathogen population was found at the center of origin of bananas which is in Southeast Asia.
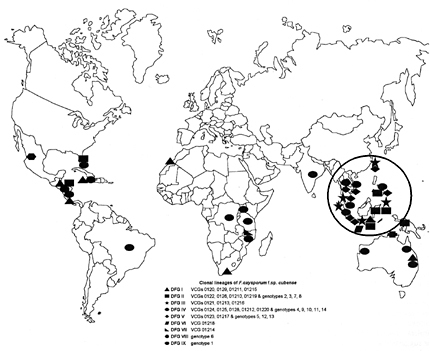 |
| Figure 10. The distribution of clonal lineages of Fusarium oxysporum f. sp. cubense on banana plantations around the world. Each symbol represents a different clonal lineage. The most diverse concentration of lineages corresponds with the presumed center of origin in Southeast Asia (circled). Click here to see an enlargement of this figure. |
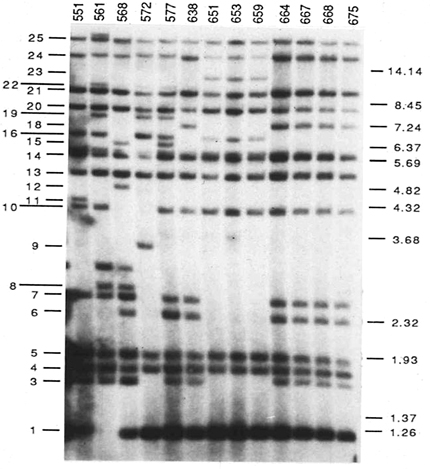
Example 3: Evidence for global movement of a single clone of the potato late blight pathogen Phytophthora infestans. DNA fingerprints (Figure 11, Goodwin et al. 1992) were used to show that the global pandemic in the 1840s was most likely due to movement of a single clone out of Mexico, which is the center of diversity and the likely center of origin of this fungus. After moving into North America, the fungus migrated on infected potatoes to Europe, and then migrated globally via trade (Figure 12, Goodwin 1997; Goodwin et al. 1994). This fungus requires two mating types for sexual reproduction. Since only one mating type escaped originally, all P. infestans populations were asexual until recently. Beginning in the late 1970s, new clones "escaped" from Mexico, including the opposite mating type and now there is increasing genotypic diversity in P. infestans populations worldwide. The metalaxyl fungicides do not work as well against the "new" populations and new populations are beginning to show signs of sexual reproduction. The first confirmation of the A2 mating type outside of Mexico was in Switzerland in 1980.
 |
|
Figure 11. DNA fingerprints based on hybridization with the probe RG57 were used to identify clones of Phytophthora infestans. |
|
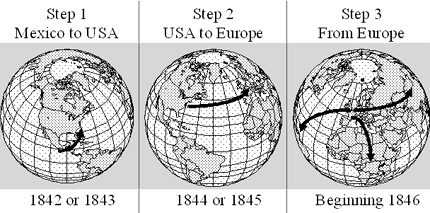 |
| Figure 12. The series of migration events that likely led to the panglobal spread of a single clone of Phytophthora infestans (Goodwin 1997). Click here to see an enlargement of this figure. |
The effects of genetic drift can be overcome by gene flow. If enough individuals are exchanged between two populations that are experiencing independent genetic drift, then the drifting populations become genetically linked and population subdivision will not occur. Sewall Wright explained this best with his population genetic parameter Nem. As before, Ne is the effective population size (a measure of genetic drift), and m is the percentage of the recipient population made up of immigrants (a measure of gene flow). The product of these two parameters, Nem, is a measure of the average number of migrants exchanged among populations each generation. A value for Nem can be estimated using a measure of population subdivision called FST, or by using private alleles, alleles found only in one population.
If Nem = 0, no migrants are exchanged between populations. The result is that different alleles can be fixed in different populations through genetic drift. Populations diverge and population subdivision occurs.
If Nem >1, meaning that on average one or more individuals are exchanged between populations each generation, then populations will not diverge by genetic drift and they will gradually become similar. Very little gene flow is needed to counteract genetic drift.
If Nem = 1, the effects of drift are exactly counterbalanced by the effects of gene flow, and the populations do not diverge or converge.
This principle is best illustrated with an example.
Assume p = q = 0.5, in other words, the two alleles at a locus are present at equal frequencies.
With Ne = 10, the effects of drift are expected to be large: Var(p) = 0.0125 (s.e. 0.11). In this population, 1 immigrant (Nem = 1) corresponds to m = 0.10; thus, 10% of the population is made up of migrants. To counteract a small Ne, m must be relatively large.
With Ne = 10,000, the effects of drift are expected to be small: Var(p) = 0.0000125 (s.e. 0.0035). In this population, 1 immigrant (Nem = 1) corresponds to m = 0.0001; thus, one-hundredth of one percent of the population is made up of migrants. To counteract a large Ne, m can be very small.
A metapopulation is a set of local populations connected by migrating individuals. The local populations may undergo repeating cycles of extinction and recolonization, while the metapopulation can remain relatively constant. A metapopulation is a population of populations.
To understand metapopulations, it helps to realize that populations are never really at equilibrium (except in mathematical models), so you can consider a species as a collection of small populations that are not at equilibrium.
Metapopulation models may offer a good representation of how pathogens evolve in agroecosystems, especially if the pathogen is a biotroph that cannot exist without a living host. In agricultural ecosystems, a new niche for a pathogen opens when a field is planted to a susceptible crop. Colonization (in this case possibly representing a founder effect) occurs when the pathogen encounters the crop. The pathogen niche is removed when the crop is harvested. After the plant host is removed, the pathogen population goes extinct or experiences a bottleneck. If the pathogen produces long-lived overseasoning survival structures, then the metapopulation model is not such a good representation.
As an example of plant pathogens that fit the metapopulation model quite well, consider the case of the cereal rusts that colonize wheat, barley, and oats each year in the "Puccinia pathway" in North America, illustrated in Figure 13.
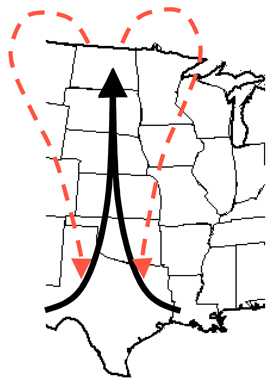 |
|
Figure 13. The Puccinia pathway in North America offers a good example of a pathogen metapopulation. Rust spores move north with prevailing winds during the spring and summer, and return to the south when prevailing winds shift direction in the fall.
|
As a result of removal of the alternate barberry host, the overwintering stage of the wheat stem rust fungus Puccinia graminis f. sp. tritici is practically non-existent in this area. Many rust fungi overwinter in the southern-most state of Texas, or in Mexico. Spores move north on the prevailing wind, following the developing cereal crops, and arrive in Canada in time to infect spring-planted cereals in the summer. The specific pathotypes that colonize each cereal field will be determined by the specific resistance genes present in the cereal cultivars grown in each field. This process will be explained further in the section on selection. During the fall when cereals are harvested in Canada and the northern USA, the prevailing wind shifts to a southerly direction and some rust spores are able to move south and infect volunteer cereal plants, thus reversing the direction of migration. The cold winters in Canada and the northern USA ensure that no spores survive the winter to begin an epidemic cycle in the following year, so the local rust population goes extinct during the winter if no alternate hosts are available. Cereal crops are recolonized by migrants from the south during the summer of the following year.
The wheat leaf rust pathogen Puccinia triticina (Puccinia recondita f. sp. tritici) reproduces only asexually in North America, so the pathogen population is composed of a series of clones and clonal lineages that move north, following the wheat crop each year.
Imagine a series of farmer's fields distributed along the Puccinia pathway. These fields are colonized by urediniospores that come from distant fields (from Southern USA or Mexico) and from neighboring fields. In each farmer's field, the local fungal population can go to extinction by:
1) harvesting the crop,
2) applying a fungicide,
3) rotating to a non-host crop,
4) planting a resistant cultivar, and/or
5) a cold winter.
After extinction has occurred, these fields can be recolonized when the farmer plants a new wheat crop. The primary inoculum that initiates the epidemic in each field can come from distant populations or from neighboring fields.
The dispersal distance and the amount of primary inoculum introduced into uninfected fields play a large role in determining the neighborhood size for the pathogen. The genetic neighborhood for a pathogen is the geographical area over which populations exchange enough migrants to evolve as a single unit.
Go to Knowledge Test for Interactions/Genetic Structure
Go to References
Next Section